国内に1人だけ、世界にも数人しか患者がいないような極めて珍しい病気がある。「超希少疾患」などと呼ばれるもので、患者が少ないことから治療薬開発はなかなか進まない。そんな状況をなんとかしたい―。ただ1人の患者のため、最新の医療技術を使って原因となっている遺伝子の変異を標的とした治療薬を作る試みが始まっている。
治療が難しい病気にどう挑み、同時に安全性をいかに確保するのか。患者や家族の願いに応えようと、通常とは異なる薬づくりに取り組む最前線の動きを探る。(共同通信=岩村賢人)
▽「核酸医薬」は有力な選択肢、病気の進行止める可能性も
日本は希少疾患を「患者数が5万人未満の病気」と定義しており、中には患者が1人というものもある。種類は約1万あるとされ、一つ一つの患者数は少ないが、全てを合わせると世界では約3億人になるとの試算がある。症状は「発達の遅れ」「代謝異常」「運動障害」「慢性的な体の痛み」などと幅広い。大半は遺伝子の異常が原因で発症し、症状は重く命にも関わる。今は遺伝子を調べる技術が発展し、原因の特定は可能になってきたが、治療法の開発は容易ではない。
こうした患者の治療に使えるかもしれない薬が「核酸医薬」だ。生物の遺伝情報を担うDNAやRNAなどの「核酸」を使用。病気の原因となるさまざまな遺伝子の変異や、異常なタンパク質に働きかける。一人一人の遺伝子変異に合わせて核酸医薬が作れれば、病気の進行を遅らせたり止めたりできるかもしれない。
筋肉の萎縮や呼吸困難が起きる難病「脊髄性筋萎縮症」の治療薬「スピンラザ」など17製品が既に日本や欧米で承認されている。製造技術は確立されており、原因となる遺伝子変異に働きかける核酸医薬の配列が分かれば、早くて2カ月で治療薬の候補が設計できる。
▽遺伝子変異発見から薬の投与まで、わずか10カ月
1人の患者を対象とした医療は「N―of―1医療」と呼ばれる。米国では臨床試験の特例という形で実際に治療が行われた。

初事例となったのは、「神経セロイドリポフスチン症」の少女ミラちゃんだ。この病気は「MFSD8」という遺伝子の変異によって脳が変性し、運動機能発達の遅れやてんかん発作、失明などさまざまな症状が起きる。日本の小児慢性特定疾病情報センターによると、欧米では1万人に1人の頻度で発症している。
臨床試験に関わった米ハーバード大ボストン小児病院のチームが2019年に発表した論文によると、ミラちゃんに症状が出始めたのは3歳の時。6歳になる前から発音の障害や視力の低下、食べ物を飲み込みにくいなどの症状が急速に進んだ。
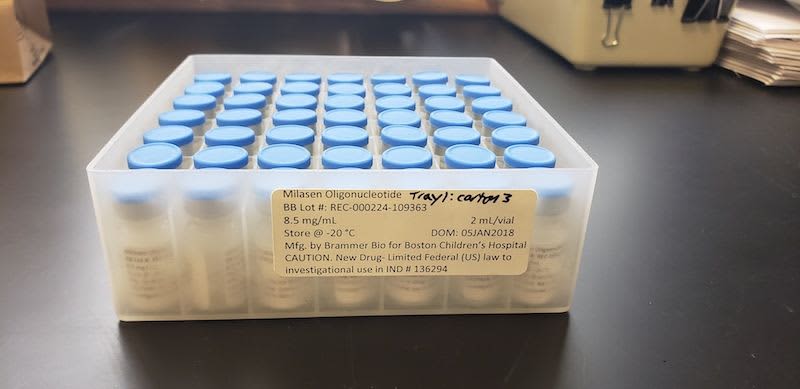
入院して検査した結果、遺伝子変異が判明したが、過去に報告がないタイプで治療薬もなかった。症状は急速に進行しており、チームは見つけた変異に働きかける核酸医薬を設計する方針を決めた。こうして作られたミラちゃんだけのための薬は「ミラセン」と名付けられた。
早急に投与するため、チームは医薬品を審査する米食品医薬品局(FDA)に働きかけ、マウスなどで安全性を検証して、短期間で臨床試験として投与する了承を得た。遺伝子変異を見つけてから投与までは約10カ月。一般の薬の開発は10年以上かかるのが普通なので驚異的な速さだった。
開発チームに所属していた中山東城・東京医科歯科大特任准教授が振り返る。「命が危ない、猶予がないという点を考慮して、FDAが迅速に認めてくれた」。核酸医薬の候補となる配列も早く見つけられた幸運なケースだった。
薬の投与を続けた結果、ミラちゃんのてんかん発作の頻度は以前と比べて50%以上も減った。米メディアによると、体を支える力も戻り、栄養補給チューブを使わなくても食事を取れるようになった。
しかしミラちゃんはその後、抑えられていた症状が再発し、10歳で亡くなった。再発した原因についてはボストン小児病院で現在も分析が続いている。
▽日本でも候補者探すプロジェクトがスタート
ボストン小児病院は、ミラちゃんのケース以降も、同じように希少疾患の患者に合わせて核酸医薬を設計する取り組みを続けている。これまで数人に、個別に作られた薬を投与した。また米製薬企業「アイオニス・ファーマシューティカルズ」の創設者は「n―Lorem(エヌ・ローレム)」という財団を設立。米国以外の国からも希少疾患の患者を募り、個別に作った薬を提供する活動をしている。

ミラちゃんの母親、ジュリア・ビタレロさんは、希少疾患の患者に個別化医療を提供する国際的なネットワークを研究者らと立ち上げた。共同通信の取材にこう語る。「病気をターゲットにするではなく、遺伝子の変異に対して薬を投与する。新しい方法で、ミラだけでなく多くの人で同じ方法が使える」
日本でも候補となる患者を探すプロジェクトが始動した。中核となっているのは東京医科歯科大や国立精神・神経医療研究センターだ。
候補となり得るのは、病名も治療法も分からない患者の遺伝子を解析する「未診断疾患イニシアチブ(IRUD)」という取り組みに参加し、診断のついた希少疾患の患者。2千人以上いる。この中から年齢や症状の重さなども踏まえて核酸医薬を作れそうな患者を探している。
中心的な役割を担う横田隆徳・東京医科歯科大教授は、原因となっている遺伝子の変異が分かれば核酸医薬で治療できるかもしれない時代になった、との見方を示す。「一人一人、有効な核酸医薬を作れるかどうかを調べ、治療可能な人をできる限り見つけたい」

▽どうやって安全性を確かめる?
国内でハードルになるのは、薬を作った後の投与までの道のりだ。通常の医薬品で実施する「臨床試験」では、大勢の患者を「薬を投与するグループ」と「投与しないグループ」に分けて比較し、有効性や安全性を確かめる。だが1人の患者向けに作った核酸医薬ではこの手法が使えない。
核酸医薬のリスクは何が想定されるのか。例えば、標的となる遺伝子に作用したものの、効き過ぎてかえって有害な反応を起こすことが考えられるという。また、標的ではない遺伝子に働いたり、予期せぬ副作用を起こしたりするケースも起こり得る。
そのため、人に投与する前に、繰り返し投与しても大丈夫か、遺伝子を傷つけないか、という点を調べる必要がある。横田さんは訴える。「通常の臨床試験が間に合わない、急速に病状が進行する患者さんを治療するため、日本に適した制度を設計する必要がある」

医薬品の審査を担当する国の組織「医薬品医療機器総合機構(PMDA)」。薬の毒性評価に関わる真木一茂・上級スペシャリストは、核酸医薬をミラセンと同様に、1人を対象とする臨床試験として投与すると想定した場合には、以下の点を考える必要があると指摘する。
動物や細胞を使った試験の成績や、既に実用化している似た核酸医薬のデータから、どれくらいの量でどんな毒性が出ると予想されるのか。投与後はどんな検査をするのが適切か。体調が悪化した際に回復は可能か―。
病気の進行が速いため、一般の薬では必須となる複数の動物や細胞を使った時間のかかる試験をある程度簡略化しなければならない。真木さんによると、安全な核酸医薬を、より早く患者に届けられるよう、臨床試験として人に投与する前にどういう毒性試験が必要なのかを現在、国の研究班で検討している。
▽臨床試験以外の選択も?仕組みづくりはこれから
ただ、N―of―1医療で使う核酸医薬は、特定の患者に投与するのが目的なので、必ずしも薬事承認を目指さない。そうなると、臨床試験という枠組みを取らなくても、適応外や未承認の薬の効果や安全性を研究として調べる「特定臨床研究」という別の仕組みや、医師の責任で投与する「未承認薬による自由診療」というやり方も選択肢になる。
いずれにせよ、どの方法が妥当なのかは日本ではまだ整理されていない。既存の仕組みで対応できるのか、新しいルールが必要なのか。厚生労働省研究開発政策課の担当者は「実際に薬を作って投与したいという事例が出てこないと議論するのは難しい」と話す。
米国では、ミラちゃんの事例を受けて、FDAが倫理面にも配慮して患者に合わせた核酸医薬を開発するための手引が2021年に作成された。あくまで「たたき台」という位置付けで、候補となりそうな薬が作れた場合、FDAが1例ずつ特例の臨床試験として投与して良いかどうかを審議している。
ミラちゃんの母親ジュリアさんは言う。「実際に治療できている子どもはまだわずか。現在の仕組みでは十分に助けられない」。米国も薬の開発体制や承認のプロセスなどさまざまな点をさらに変えていく必要があると訴えている。
▽薬に見いだした希望「一番大事なのは命」
仕組みが整っていない中、核酸医薬の開発に希望を見いだしている患者や家族がいる。
埼玉県に住む中学1年の池田陽平さん(12)=仮名=は、2歳になった頃からふらついてよく転ぶようになった。母親は「支えがないとうまく座れない。よだれがよく出ていたことも気になった」と振り返る。整形外科などいくつかの病院を回ったが原因は分からぬまま。4歳の時に国立精神・神経医療研究センターで遺伝子を調べて「毛細血管拡張性運動失調症(A―T)」と診断された。
11番染色体にある遺伝子「ATM」の変異によって脳と免疫に影響が出る病気だ。全身の筋肉をうまく調節できず、ふらついたり、手をうまく動かせなくなったりする。感染症で重症化しやすい、がんを発症しやすい、などの症状も起きる。患者会によると、発症するのは人口10万人当たり1人の割合。どう症状がどう変化していくのか予想するのが難しく、治療法は確立されていない。
診断後、免疫の働きを補う薬を週1回、専用の機器で注射している。それでも症状は少しずつ進行し、小学3年の頃から手が震えてうまく字が書けなくなった。長く同じ姿勢で座るのが難しくなり、飲み込む力が落ちていて食後に吐いてしまうこともある。現在は特別な車いすで学校に通っている。
母親は「小学4年生ぐらいまでは基本的には元気で、感染症で入院するという経験もなかった」と話す。しかし、その後、血液がんになる可能性が高いと分かって骨髄移植をし、疲れやすいため自宅で過ごす時間の多くは横になっているという。
個別に作った核酸医薬を使えば、症状の進行を遅らせて、より長く生きられる可能性がある。一方で、根治させたり、失われた体の機能を回復させたりするのは難しい。
手を引いてもらえば歩けるようになるとか、少しでも体の不自由がなくなることを本人は一番望んでいるのではないか―。母親はそう考えており、作られた薬にどんな効果がありそうかを知った上で、投与するかどうかを陽平さんと一緒に考えるつもりだ。生きていれば、いずれ画期的な治療法が開発されて、受けられるかもしれない。「一番大事なのは命。できるだけ長く生きていてほしい。早く薬を作って投与できるなら、それが望ましいと思う」

